SMA er en sjælden genetisk neuromuskulær sygdom, der påvirker den del af nervesystemet, som kontrollerer frivillig muskelbevægelse.1
HVAD ER SPINAL MUSKELATROFI (SMA)?
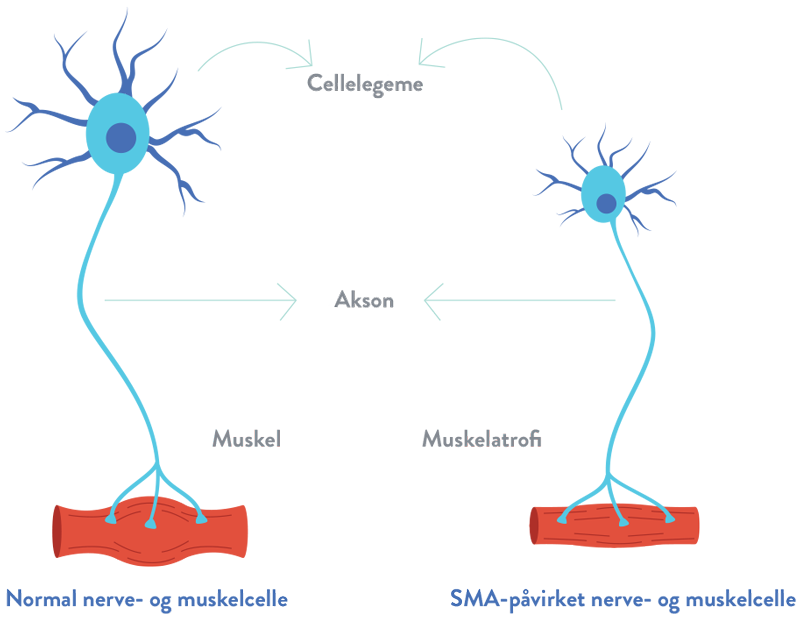
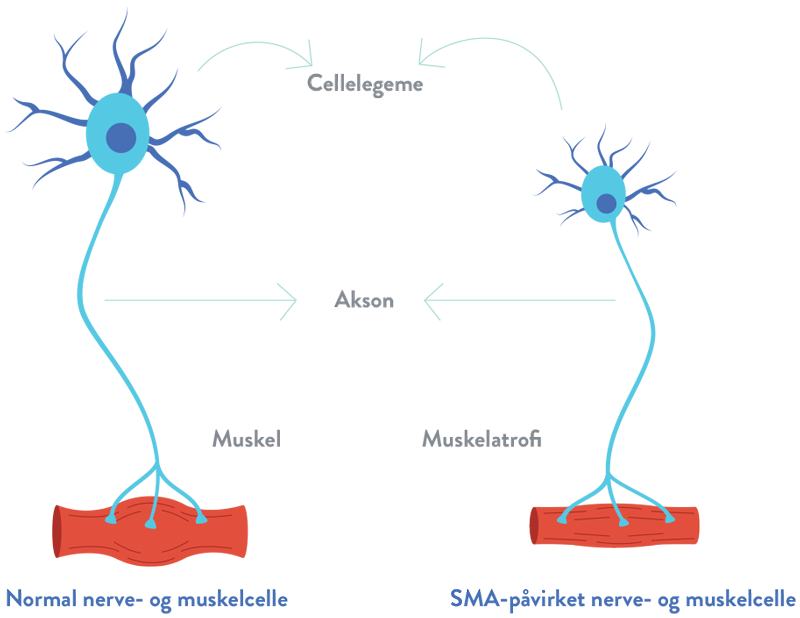
Ved spinal muskelatrofi (SMA) sker der tab af vigtige celler i rygmarven, kaldet motorneuroner, som er vigtige for muskelstyrke og bevægelighed. Disse motorneuroner regulerer muskelaktiviteten ved at sende signaler fra det centrale nervesystem (CNS), som er den del af kroppens nervesystem, der omfatter hjernen og rygmarven.1,2
Tabet af fungerende motorneuroner fører til tiltagende muskelsvaghed og atrofi (gradvis reduktion af musklernes masse og styrke), efterhånden som musklerne holder op med at modtage signaler fra CNS.3
I modsætning til mange andre sjældne neuromuskulære sygdomme har man en klar forståelse af den specifikke genetiske årsag til spinal muskelatrofi (SMA).
HVAD ER ÅRSAGEN TIL SPINAL MUSKELATROFI (SMA)?
Spinal muskelatrofi (SMA) er forårsaget af en mutation i genet SMN1 (survival motor neuron 1). Dette gen er ansvarligt for produktion af SMN-protein, som holder motorneuronerne intakte og sikrer, at de fungerer normalt.
Hos mennesker med spinal muskelatrofi (SMA) er begge kopier af SMN1-genet muterede, og det fører til nedsat produktion af SMN-protein. Uden et tilstrækkeligt niveau af SMN-protein, vil motorneuroner i rygmarven gå tabt og det forhindrer musklerne i at modtage korrekte signaler fra hjernen.4,5
Degenereringen af motorneuroner fører til den gradvise reduktion af musklernes masse og styrke (atrofi).
Kun til illustration.
Kun til illustration.
HVILKEN KONSEKVENS HAR SPINAL MUSKELATROFI (SMA) FOR ET BARN?
Børn med spinal muskelatrofi (SMA) påvirkes forskelligt, og det er vigtigt at bemærke, at symptomerne kan variere meget afhængigt af barnets alder ved sygdommens frembrud samt sygdommens sværhedsgrad. Børn kan opleve fremadskridende muskelsvaghed i de muskler, der sidder tættest på kroppens centrum, såsom skuldre, lår og bækken. Disse muskler muliggør aktiviteter såsom at kravle, gå, sætte sig op og styre hovedets bevægelser. Åndedræt og synkefunktion kan også blive påvirket.7
Spinal muskelatrofi (SMA) påvirker ikke de neuroner, der er ansvarlige for kognition, som er den mentale proces, hvorved vi får viden og forståelse via tanker, erfaringer og sanser.8,9 Ifølge en undersøgelse har børn og unge med spinal muskelatrofi (SMA) normal intelligens med IQ i standardområdet. Intelligenstest samt kognitiv og adfærdsmæssig test kan bidrage til, at børn i skolealderen ikke keder sig, er understimulerede eller frustrerede.10
HVAD BØR JEG VIDE OM SMN2?
Alle mennesker med spinal muskelatrofi (SMA) har mindst et "backup-gen", kendt som SMN2. SMN2-genets struktur ligner strukturen SMN1, men kun en lille mængde (10 %) af det SMN-protein, det producerer, er fuldt funktionelt. Dette lave niveau af SMN-protein er ikke effektivt nok til, at motorneuroner kan overleve i CNS.11-13
Antallet af SMN2-gener kan variere, og et højere antal SMN2-kopier er forbundet med mindre alvorlige symptomer på spinal muskelatrofi (SMA).4,13,14 Sygdommen medfører en lang række symptomer, og selvom der er en stærk sammenhæng mellem SMN2-kopier og sygdommens sværhedsgrad, findes der undtagelser. Eksperter anbefaler derfor, at beslutninger om pleje træffes på basis af barnets funktionsevne og ikke antallet af SMN2-kopier. Imidlertid bruges antallet af kopier af SMN2 som et nøglekriterium for inklusion i kliniske forsøg.2,15
ER SPINAL MUSKELATROFI (SMA) GENETISK?
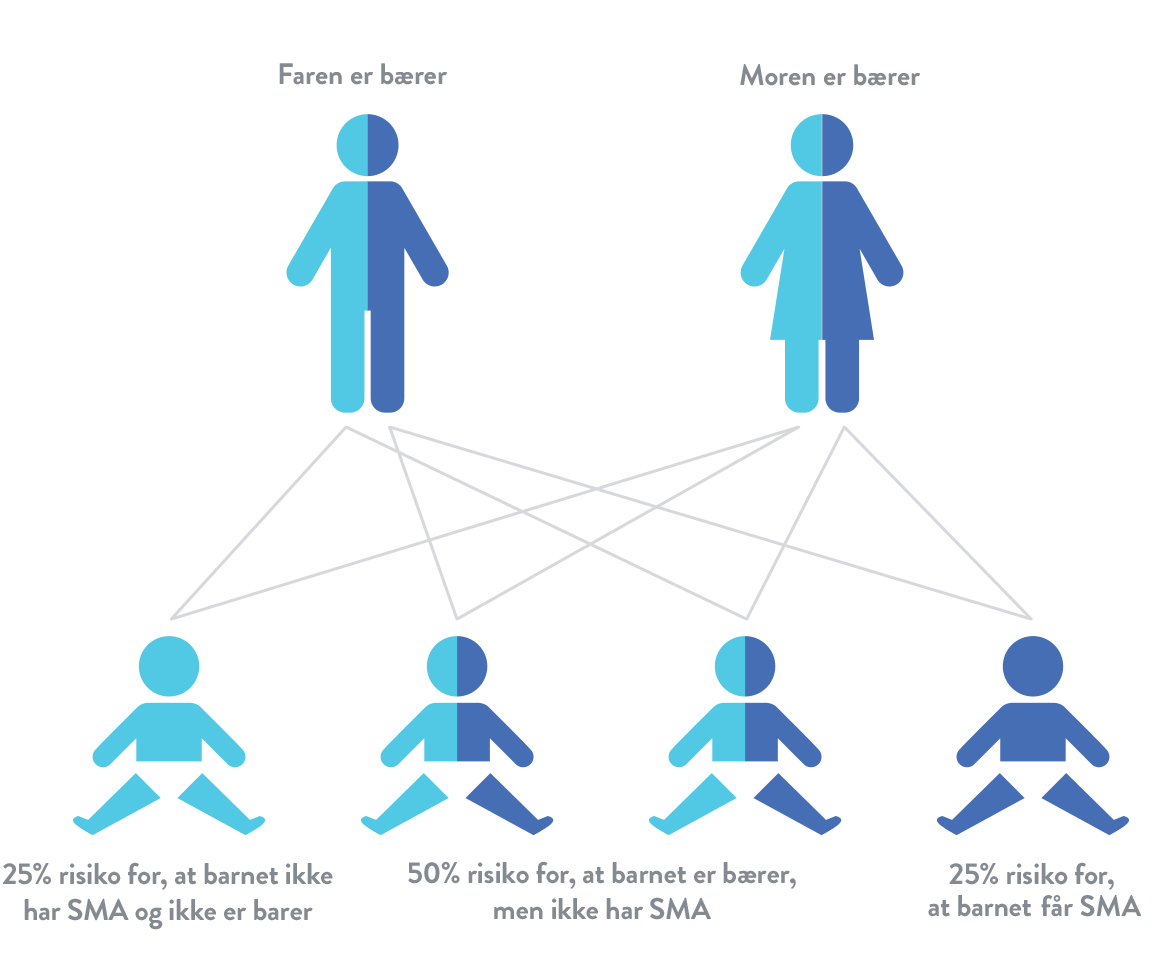
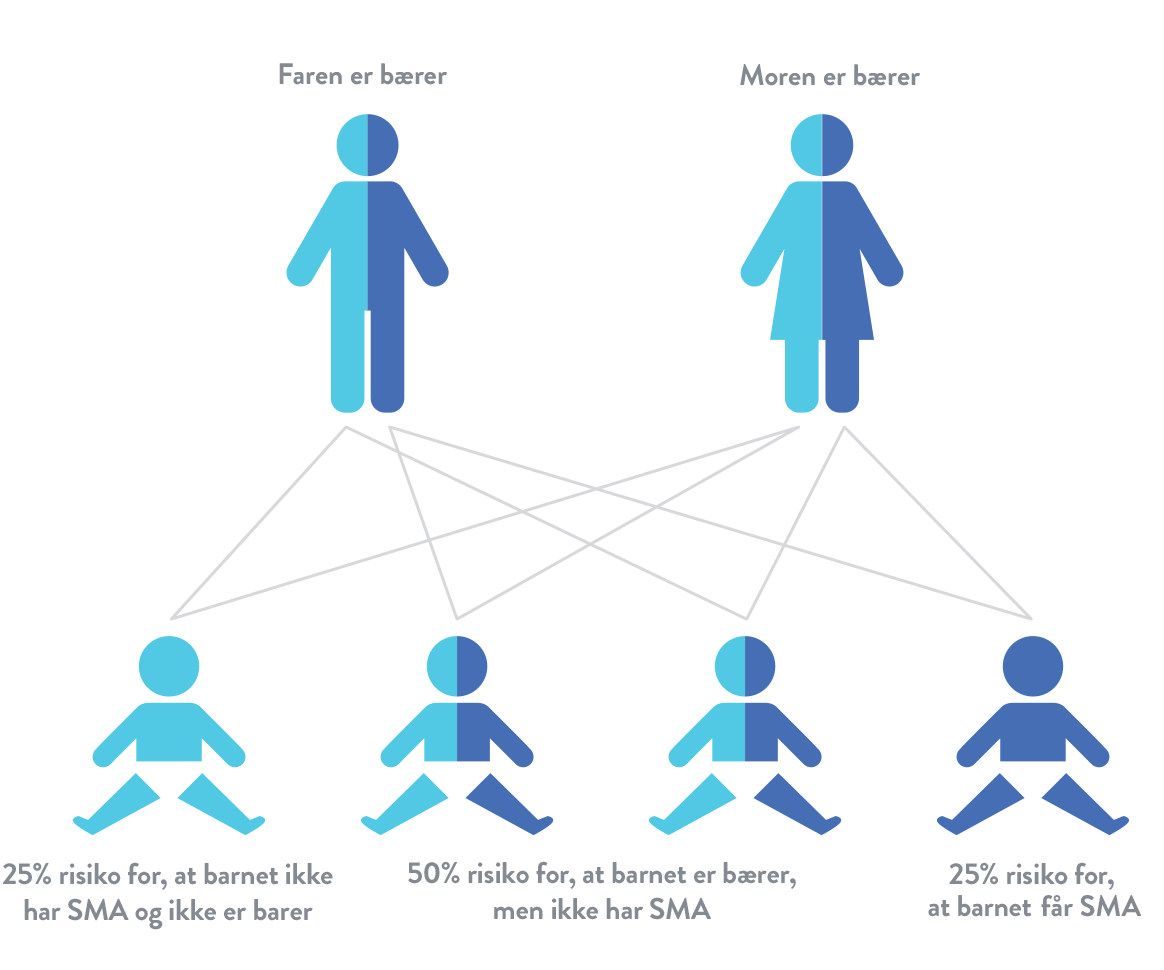
Spinal muskelatrofi (SMA) er en autosomal recessiv sygdom. Det betyder, at et barn kun er i risikogruppen, hvis det arver et muteret SMN1-gen fra hver af forældrene. Hvis et barn kun arver ét muteret SMN1-gen, betragtes det som "bærer", men har normalt ikke symptomer på spinal muskelatrofi (SMA).16
Hvis to bærere af et muteret SMN1-gen får et barn, er risikoen:
Hvis der er spinal muskelatrofi (SMA) i din familie, er din risiko for at være bærer større end gennemsnittet. Når du skal beslutte, om du vil have børn, kan det være nyttigt at rådføre dig med din læge for at få at vide, hvilke mutationer der er almindelige i din familie. Når din families mutation er kendt, kan en test, der passer til din situation, fastlægges:
Hvis din families mutationer er SMN1-deletioner, kan test af kopiantal være passende for dig
Hvis din families mutationer omfatter en mere subtil ændring i genet, kan din læge og laboratoriet muligvis beslutte, om der kan udføres test for at finde den specifikke ændring
Hvis du ikke kan få oplysninger om din families mutationer, kan du alligevel godt få udført en test af kopiantallet. Din risiko for at være bærer (inden du bliver testet) beregnes ud fra din familiehistorie. Hvis dine resultater er normale, kan din risiko for at være bærer være mindre.
Mange laboratorier og hospitaler tilbyder bærerscreening for at bestemme, om den ene eller begge forældre er bærere af det muterede SMN1-gen. Dette kan give enkeltpersoner og familier oplysning om risikoen for at få et barn med spinal muskelatrofi (SMA).17 En medicinsk genetisk rådgiver er uddannet til at gøre oplysninger om genetiske risici, test og diagnose lettere for familier at forstå.